
.
| ∴ | Zusammenfassung (Abstract). Untersucht wird ein Quantenalgorithmus zur Modellierung von Wechselwirkungen der Atome innerhalb eines Moleküls, vorgestellt durch Schütt, Gastegger, Tkatchenko, Müller, Maurer 2019 in „nature“; im folgenden „SGTMM-Algorithmus“. | ∴ |
.
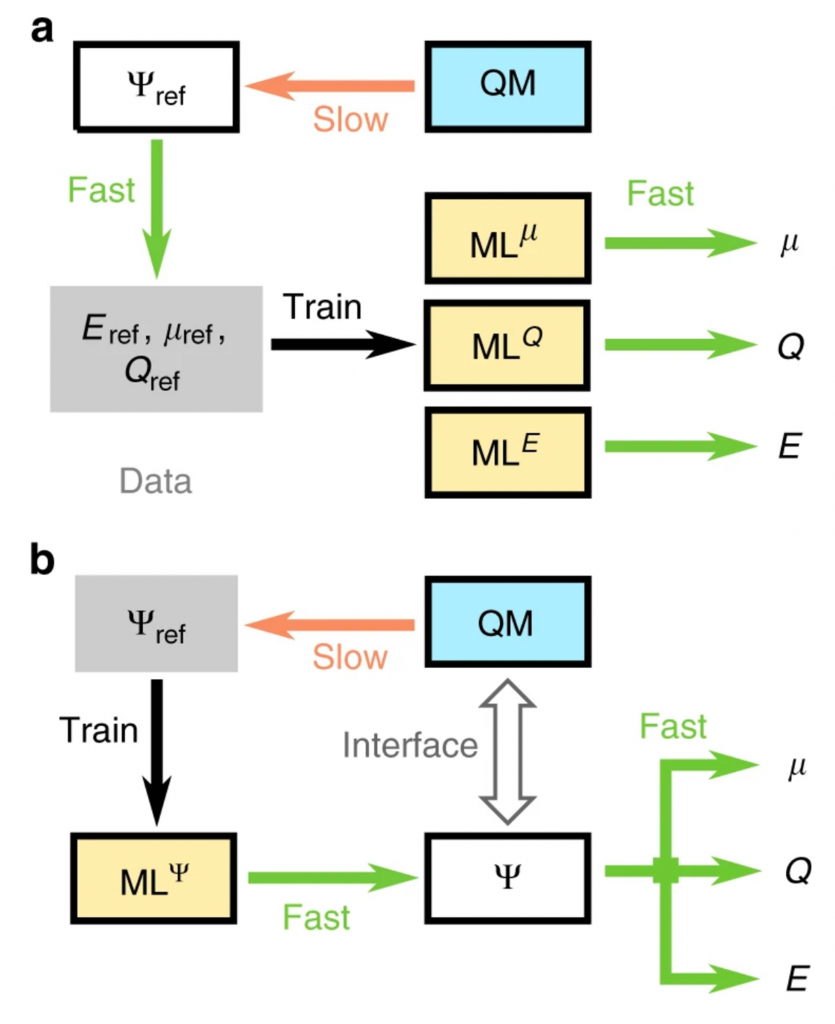 Knappe 3 Jahre ist es her, als ich über die Fortschritte von Rainer Blatt’s Qubit-Forschung und der damit verbundenen Hoffnung auf ganz neue Werkstoffe in dem nachfolgend verlinkten
Knappe 3 Jahre ist es her, als ich über die Fortschritte von Rainer Blatt’s Qubit-Forschung und der damit verbundenen Hoffnung auf ganz neue Werkstoffe in dem nachfolgend verlinkten ![]() – Post berichtete. Dies hat zunächst die üblichen Bedenkenträger auf den Plan gerufen: „braucht kein Mensch“, „zu teuer“, „geht auch einfacher“ und ja und überhaupt 🙄. Dann tauchten darunter aber auch andere Stimmen auf, die neben der verständlichen Frage, wie das denn funktionieren solle, eben die Kernfrage stellten: warum schafft es ab 36, 40 oder wieviel auch immer Atomen im Molekül nur der Quantencomputer? Anders ausgedrückt, warum liegt die Grenze zur „quantum primacy“ ausgerechnet bei dieser Größenordnung von Qubits im Quantenregister? Genau dieser Fragestellung widmet sich die vorliegende AG.
– Post berichtete. Dies hat zunächst die üblichen Bedenkenträger auf den Plan gerufen: „braucht kein Mensch“, „zu teuer“, „geht auch einfacher“ und ja und überhaupt 🙄. Dann tauchten darunter aber auch andere Stimmen auf, die neben der verständlichen Frage, wie das denn funktionieren solle, eben die Kernfrage stellten: warum schafft es ab 36, 40 oder wieviel auch immer Atomen im Molekül nur der Quantencomputer? Anders ausgedrückt, warum liegt die Grenze zur „quantum primacy“ ausgerechnet bei dieser Größenordnung von Qubits im Quantenregister? Genau dieser Fragestellung widmet sich die vorliegende AG.
Leser, die an dieser Stelle womöglich fragen werden, ob es denn nicht eine Nr. kleiner geht, kann ich wohl ein wenig beruhigen. Denn auch diese AG hat – wie jede andere „AG Energetik“ auch – unverändert den Zweck der Vermittlung von Methoden der Erkenntnisgewinnung…💡 Außerdem stützen wir uns hier auf einen bereits fertig konzipierten Algorithmus, der kürzlich in der Zeitschrift „Nature“ unter »https://www.nature.com/articles/s41467-019-12875-2« vorgestellt worden ist. Wir nennen es „SGTMM-Algorithmus“ (nach den Namen dessen Urheber); das Paper der Studie ist im Anschluss an die AG eingebettet. Mit anderen Worten, wir erfinden hier keinen modifizierten oder gar neuen Quantenalgorithmus, sondern betrachten vielmehr bekannte Sachen unter einem anderen Blickwinkel; vgl. Tutorial zu Deutsch-Jozsa-Problem.
.
.
https://www.facebook.com/rainer.stawarz/posts/1821870624695484
.